Drug Metabolism and Transport
Metabolism of Small Molecule Drugs
Molecular weight < 1000 g/mol (includes most oral drugs)
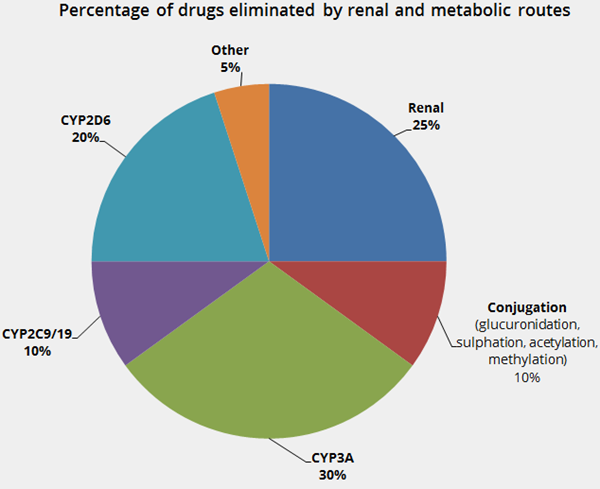
- Responsible for the elimination (deactivation) of 70% of drugs - see the
 Drug Elimination Pie Chart.
Drug Elimination Pie Chart. - Responsible for the activation of a few drugs i.e. prodrugs.
- Most drugs are predominantly metabolised by cytochrome P450 enzymes (CYPs). Most of the remaining drugs are predominantly metabolised by conjugating enzymes (UDP-glucuronosyltransferase enzymes (UGTs), esterases, and sulfotransferase enzymes (SULTs)).
- Enzyme function can be increased (induced) or reduced (inhibited) by disease, drugs, or genetics.
- Drugs may be metabolised by more than one enzyme. Usually one enzyme contributes the most to metabolism.
- Enzyme inhibitor – decrease the function of metabolising enzymes. Drugs metabolised by these enzymes have reduced clearance, higher plasma concentrations and greater clinical effect. This can cause toxicity.
- Can affect a single enzyme or several.
- Onset of inhibition is usually immediate.
- The time to full effect (or loss of effect) varies depending on the half lives of both drugs and the mechanism of inhibition (sometimes irreversible). Maximum effect is usually reached in hours to days (rather than minutes or weeks).
- Enzyme inducers - increase the function of metabolising enzymes. Drugs metabolised by these enzymes have increased clearance, lower plasma concentrations and reduced efficacy. This can cause therapeutic failure.
- Usually affects multiple enzymes.
- Onset of induction is slower than inhibition.
- The time to full induction or recovery from induction is approximately one week.
- Prodrugs are inactive drugs metabolised to the active form by an enzyme. Inhibition can cause loss of efficacy and induction can cause toxicity.
- The metabolism tables contain lists of common substrates of the various enzymes, and major inhibitors and inducers.
Metabolism of Large Molecule Drugs
Molecular weight > 1000 g/mol (e.g. monoclonal antibodies and insulin)
- Eliminated (deactivated) by degrading enzymes (proteases) and/or by binding irreversibly to a “target” molecule.
- Clearance varies many-fold between individuals.
- Pharmacokinetic drug-drug interactions are not usually a problem.
Saturable Metabolism
- Saturation of metabolism occurs when enzymes involved in the metabolism of drugs have reached capacity.
- Most drugs do not have saturable metabolism at standard doses.
- Concentration changes in proportion to maintenance dose, e.g. double maintenance dose leads to doubling in steady-state concentrations.
- A few drugs have saturable metabolism.
- Disproportionate concentration changes occur, e.g. phenytoin – a double maintenance dose can lead to a many-fold increase in steady-state concentrations.
- In overdoses, saturable metabolism can occur for many drugs.
Drug Transport
- Important for absorption, distribution and elimination of drugs, and for drugs that act on intracellular targets.
- At steady-state, drug transporters create a concentration gradient across a cell membrane.
- Drug transporters move drugs across cell membranes. There are three main types:
- Efflux transporters – these remove lipophilic drugs out of cells and the body.
- Solute transporters – these transport ions (anion and cation) including hydrophilic drugs in and out of cells and the body.
- Vesicular transporters – these move large molecules across cell membranes by endocytosis and exocytosis.
- Genetic polymorphism exists.
- Drugs can be substrates of more than one transporter.

Topic Code: 93324